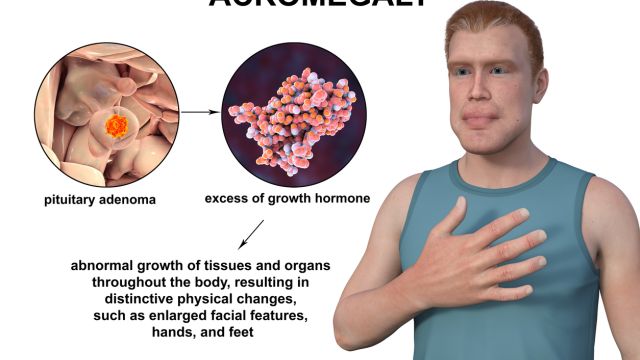
Akromegalia jest chorobą o charakterze przewlekłym nadmierne wydzielanie hormonu wzrostu (GH). Wysoki poziom hormonu wzrostu powoduje gigantyzm przy niepełnym wzroście, a po zamknięciu stawów nasadowych – akromegalię. Akromegalia jest rzadką chorobą, występującą z częstością od 40 do 70 przypadków na milion osób i roczną zapadalnością na poziomie od 3 do 4 nowych przypadków na milion osób.
Pulsacyjne wydzielanie hormonu wzrostu przez komórki somatotropowe gruczolakowatości przysadki jest kontrolowane przez czynnik uwalniający hormon wzrostu (GHRH), który ma działanie stymulujące i hamowane przez somatostatynę. Grelina, wydzielana głównie z dna żołądka, stanowi dodatkowy, choć słaby, bodziec do wydzielania hormonu wzrostu. U zdrowych ludzi występuje hormon wzrostu wydzielanie pulsugłównie podczas snu wolnofalowego lub podczas ćwiczeń.
U ponad 95% akromegalia występuje w wyniku przewlekłego narażenia na nadmiar hormonu wzrostu wydzielanego przez łagodny nowotwór – gruczolak przysadki mózgowej, pochodzące z komórek somatotropowych. Jednakże u mniejszości pacjentów występuje rodzinna lub syndromowa akromegalia. Obejmuje rodzinny izolowany gruczolak przysadki, mnogą neoplazję endokrynną 1 i 4, akrogigantyzm sprzężony z chromosomem X, złożony Carneya, dziedziczny przyzwojak, guz chromochłonny i nerwiakowłókniakowatość 1. Większość przypadków akromegalii wynika ze sporadycznych gruczolaków. W bardzo rzadkich przypadkach akromegalia może być spowodowana ektopowym wydzielaniem hormonu uwalniającego hormon wzrostu (GHRH) lub PX.
Hormon wzrostu wywiera wielostronny wpływ na metabolizm i tkanki. W pośrednim działaniu hormonu wzrostu pośredniczą stymulacja insulinopodobnego czynnika wzrostu 1 (IGF-1) z hepatocytów, komórek mięśniowych i kostnych. U zdrowych osób wydzielanie hormonu wzrostu znajduje się pod kontrolą ujemnego sprzężenia zwrotnego z krążącym IGF-1, z którego większość pochodzi z wątroby. W przeciwieństwie do krążącego hormonu wzrostu, poziomy IGF-1 w surowicy są stabilne w ciągu 24 godzin i służą jako dokładny pomiar działania hormonu wzrostu.
W akromegalii dochodzi do utraty normalnego, pulsacyjnego wydzielania hormonu wzrostu, a jego poziom trwale wzrasta. Nie ma charakterystycznego nocnego spadku, a reakcje na stymulację i tłumienie są nieprawidłowe. W tym przypadku glukoza nie działa hamująco na hormon wzrostu i nie powoduje stymulacji hipoglikemii. Przewlekłe nadmierne wydzielanie hormonu wzrostu następuje poprzez stymulację IGF-1, co prowadzi do: proliferacja kości, chrząstek, tkanek miękkich i wzrostu narządów wewnętrznych.
Obraz kliniczny pacjentów z akromegalią zależy od działania hormonu wzrostu i insulinopodobnego czynnika wzrostu (IGF-1). Zmiany obserwuje się m.in wiele narządów i układów.
Zmiany kostne – zwiększenie szerokości nadgarstków i stóp, zgrubienie rysów twarzy, pogrubienie sklepienia czaszki, wydłużenie i poszerzenie żuchwy, prognacja, diastema. Zmiany te wymagają zmiany ilości butów, rękawiczek.
Skóra i tkanki miękkie – szorstka skóra z mięśniakami, nadmierne owłosienie, wzmożona potliwość i łojotok, powiększenie nosa, warg, małżowin, języka, pogrubienie strun głosowych.
Wisceromegalia – ze względu na anaboliczne działanie hormonu wzrostu wpływa na różne narządy – kardiomegalię, powiększenie śledziony, powiększenie wątroby, tyromegalię, renomegalię. U kobiet – macica i powstawanie węzłów chłonnych.
Kifoskolioza, powiększenie przednio-tylnej średnicy klatki piersiowej, powiększenie płuc, deformacja tchawicy, niedrożność górnych dróg oddechowych, bezdech senny. U takich pacjentów może być konieczna tracheostomia w trybie nagłym.
Nadciśnienie tętnicze, kardiomiopatia. Powikłania sercowo-naczyniowe są najczęstszą przyczyną zgonów.
Objawy neurologiczne – parestezje kończyn, objawy cieśni nadgarstka, depresja, senność.
Inne zaburzenia endokrynologiczne – guzek wola, nadczynność tarczycy, zmniejszona tolerancja węglowodanów w przebiegu ciężkiej cukrzycy, obniżone libido, zaburzenia miesiączkowania.
Występuje zwiększona tendencja do polipów w przewodzie pokarmowym i nowotworów – najuczciwiej w okrężnicy, przełyku, żołądku, piersi.
Objawy wynikające z ucisk nowotworu – ból głowy, pogorszenie widzenia, zwężenie pola widzenia, wtórny hipogonadyzm, niedoczynność tarczycy, hipokortykalizm. Choroba rozwija się stopniowo przez dziesięciolecia, a od pierwszych podejrzanych objawów do postawienia diagnozy mija zwykle 7–10 lat.
W plan diagnostyki różnicowej enter – cecha rodzinna członków rodzin o grubszych rysach, pachydermoperiostozie i silnie wyrażonym zespole insulinooporności. W tych chorobach wydzielanie PX i IGF-1 jest normalne.
Bibliografia:
Mayo Clin Proc. 2022;97(2):333-346
Zalecenia dobrej praktyki klinicznej w chorobach przysadki mózgowej